Rubber Elasticity
Rubber bands are made from polymers, but the chains are crosslinked to provide a network.
The amorphous phase in PE is also said to be rubbery – it is above its Tg but is constrained by the surrounding crystals and so cannot be said to be liquid-like.
For the rubber bands, it is the crosslinks which determine the properties.
[We will see later what the analogy is in amorphous regions of uncrosslinked materials.]
The crosslinks provide a 'memory'.
When the network is stretched, entropic forces come into play which favour retraction, returning the network to its original unstretched/equilibrium state.
Changes to the Rubber Network upon stretching
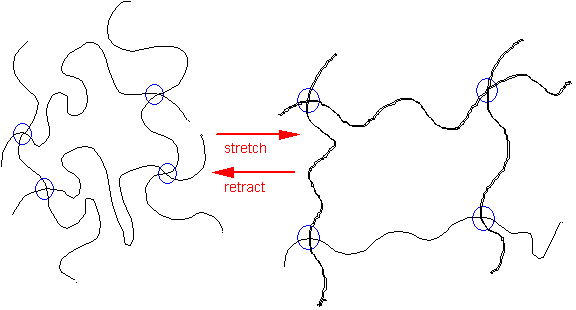
Loss of entropy upon stretching, means that there is a retractive force for recovery when external stress removed.
This is why a rubber band returns to its original shape.
Use statistical mechanics to provide equations for the force on the chains.
Consider a 1D random walk:
Step length p
Total distance travelled x
N steps of which A are forward and B back
Then A+B = N and (A-B)p = x
Total distance travelled can be achieved in W ways where W = ![]()
Can use Stirling's approximation and solve for A andB in terms of N, p and x.
Then ln W = N ln 2
- 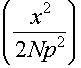
And therefore S = Nkln2
- 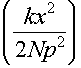
This can be generalised to the 3d case
 |
S = 3Nkln2 - 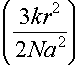
where r is total distance and a is step length in 3d
Can now use thermodynamics to relate change of entropy to tension on a single chain when deformation applied
Change in free energy when external force f applied
fdx = d(U – TS)
Þ
f = 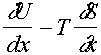
bond distortion term:
usually negligible
f = 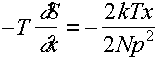
 |
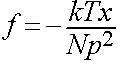 for 1D case
for 1D case
i.e behaves as a classical spring of zero unstrained length – Hookeian elasticity.
Network Elasticity
However each chain does not deform individually but is part of a network.
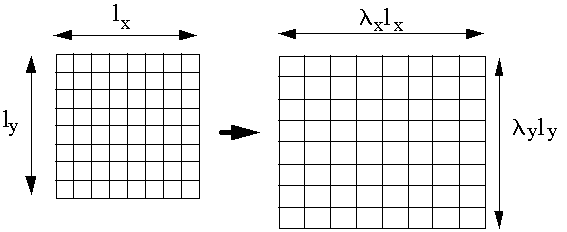
Initial (3D) vector ro = ( x, y, z) between two crosslink points deforms to r = (lxx, lyy, lzz)
i.e r2 = lx2x2 + ly2y2 + lz2z2
and Dr2 = (lx2-1)x2 + (ly2-1)y2 + (lz2-1)z2
so the change in entropy of this unit of the network is

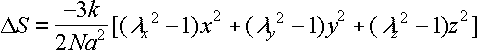
This needs to be summed over chains – n/unit vol
For an initially isotropic network
<x2> = <y2> = <z2> = 1/3 Na2
 |
Then
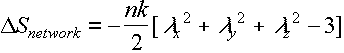
i.e depends on chain ('strand') density, but not on contour length ÖN a
Consider special case of extension in x direction
i.e lx= l and ly=lz by symmetry
Since rubber is essentially incompressible
ly=lz= (l)-1/2
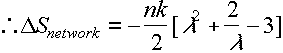
Now ignoring bond distortion
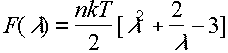
From this equation can obtain the stress-strain (or equivalently force-extension) relationship.
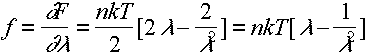
For small deformations, strain e = l-1 and 1/l ~(1+e)-1
\ f = nkT [1 + e- (1+e)-2] = 3nkTe
Since n is number of chains/unit area, this is also equal to force/unit area = stress s
\s = 3nkTe i.e. Hookeian spring behaviour
Can write this as s = Ee where E is Young's modulus
E= 3nkT
For incompressible materials, Young's modulus E= 3G (shear modulus)
G = nkT
Note this means that for entropic elasticity (unlike enthalpic) the modulus increases with temperature and the material gets stiffer rather than softer.
Since n = no of strands/unit vol, can also write this in terms of the average MW between crosslinks Mx.
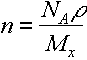 where r is the density
where r is the density
 |
\ 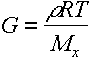
This is useful for other purposes as we will see later.
Note as cross link density goes up, (n, or equivalently Mx decreases), modulus goes up : a highly crosslinked rubber is stiffer than a lightly crosslinked one.
Limitations of model: only works for small strains - recall Stirling's approximation was used; bond distortion ignored.
Network has limited extensibility. Can improve on model using Langevin function as in the case of magnetism.
At large strains may have crystallisation occurring : strain induced crystallisation. This can occur as chains line up during extension.
Orientation
We have seen for rubbers how the presence of crosslinks leads to a memory effect. A similar sort of effect can be seen for glassy amorphous polymers, only now the polymer must be warmed up to allow retraction.
· What is the memory effect here (no crosslinks)?
· Why is orientation useful?
In glassy polymers such as PS, there are no chemical crosslinks.
However the chains are long and get all tangled up.
They behave as if there are local topological constraints – known as entanglements.
Entanglements are not permanent, and can be broken by deformation, but they do act to form a temporary network.
Chains can be stretched between entanglement points, just as with crosslinks in a rubber.
Imagine taking a glassy polymer above Tg, and then cooling down while chains still stretched.
Recall rubber stress-strain curve with the modulus greater at higher strains, as orientation of the chains occurs.
This is true for glassy polymers too.
Oriented polymers will be stiffer along the chain direction than unoriented, and much stiffer than in transverse direction. This is because there is little additional slack to pull out in the chain.
There is strong commercial drive to produce high modulus fibres which are essentially stretched out chains of PE etc.

For crystalline polymers, orientation will obviously change the type of crystals present – chain folding no longer likely.
During processing, orientation often introduced, thereby destroying existing spherulites etc.
Polymer Fracture
Glassy polymer fracture is typically brittle, but differs substantially from other fractures, by virtue of the long polymer chains.
Cracks do form, but prior to that a unique kind of deformation called crazing also occurs.
Crazes form by drawing out chains from undeformed polymer until they are essentially stretched taut between entanglement points.
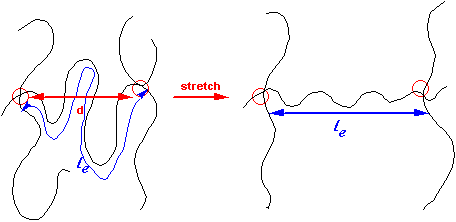 |
At this extension – which will be a characteristic for each polymer depending on the density of entanglements – further extension is difficult ('strain hardening') and the craze is stabilised.
Easy to stretch until an extension ratio
l = le/d is reached.
This can lead to extensions of up to ~4 in the craze.
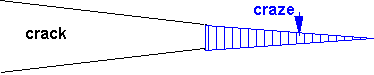 |
The craze runs ahead of a crack, and resembles it but consists of load bearing fibrils which span the interfaces.
The craze is therefore load bearing, but is a source of weakness.
Crazes can be seen in the 'stress whitening' of flexed perspex rulers.
Since each polymer has a different l (based on chain dimensions), we expect this to be reflected in craze structure.
l in the craze can be determined by electron
microscopy.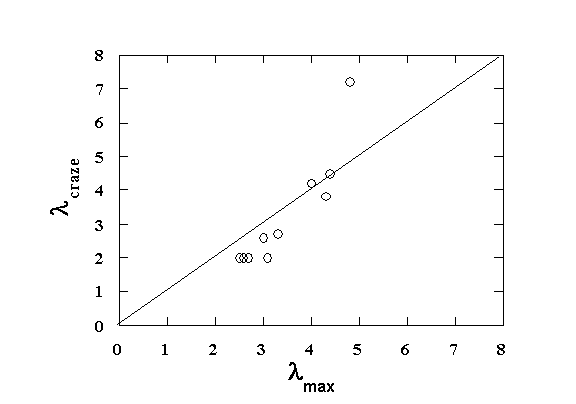
Good correlation is found.
In fact, the idea of entanglements was originally conceived for polymer melts, and the fact that these ideas could be extended to glasses – where chain motion is not expected to occur – was originally not accepted by theorists.