Phase Diagrams and Phase
Separation
Books
MF Ashby and DA Jones, Engineering Materials Vol 2, Pergamon
P Haasen, Physical Metallurgy,
G Strobl, The Physics of Polymers, Springer
Introduction
Mixing two (or more)
components together can lead to new properties:
Metal alloys e.g. steel,
bronze, brass….
Polymers e.g. rubber toughened systems.
Can either get complete mixing on the atomic/molecular level, or phase separation.
Phase Diagrams allow us to map out what happens under different conditions (specifically of concentration and temperature).
Free Energy of Mixing
Entropy of Mixing
nA atoms of A
nB atoms of B
Total atoms N = nA + nB
Then Smix = k ln W
=
This can be rewritten in terms
of concentrations of the two types of atoms:
nA/N = cA nB/N
= cB
and using Stirling's
approximation
Smix = -Nk (cAln cA + cBln
cB)
This is a parabolic curve.
There is always a positive entropy gain on mixing (note the logarithms are negative) – so that entropic
considerations alone will lead to a homogeneous mixture.
The infinite slope at cA=0
and 1 means that it is very hard to remove final few impurities from a mixture.
This is the situation if no
molecular interactions to lead to enthalpic contribution to the free energy
(this corresponds to the athermal or ideal mixing case).
Enthalpic Contribution
Assume a coordination number
Z.
Within a mean field
approximation there are
nAA bonds of A-A type = 1/2 NcAZcA
= 1/2 NZcA2
nBB bonds of B-B type = 1/2 NcBZcB
= 1/2 NZ(1-cA)2
and nAB bonds of A-B type = NZcA(1-cA)
where
the factor 1/2 comes in to avoid double counting and cB = (1-cA).
If
the bond energies are EAA, EBB and EAB
respectively, then the energy of interaction is (writing cA as
simply c)
1/2 NZ [cEAA + (1-c)EBB + c(1-c) (2EAB
- EAA – EBB)]
energy of 2 separate starting materials
\ Umix
= 1/2 NZ c(1-c) (2EAB – EAA – EBB)
The
term (2EAB – EAA – EBB) determines whether
mixing or demixing occurs.
Define ckT = Z/2 (2EAB –
EAA – EBB)
where c is known as the interaction
parameter, and is dimensionless.
(The
definition of c in this way, including the kT term, is for historical
reasons).
Fmix/ kT = c ln c + (1-c) ln (1-c) + c kT
c(1-c)
Three
cases to consider
Þ
c=0 athermal or
ideal solution case.
Fmix is always negative.
Þ
c>0 i.e. 2EAB> EAA + EBB
2
local minima result
Þ
c< 0
One
deep minimum
These
curves correspond to the case for one particular T.
In
order to plot out a phase diagram, we need to be able to see how temperature
affects these curves.
And we need to know whether or not these curves imply phase separation.
Finally
we need to know, if phase separation occurs, how much of each phase is present.
Lever Rule for Determining
Proportions of Phases
Imagine we have a system with 2 phases present (labelled 1 and 2), and two types of atoms A and B.
In phase 1 concentration of A = c1
In phase 2 concentration of A = c2
and that there is x of phase 1 present
and (1-x) of phase 2 present.
Total of N atoms, overall concentration of A = c
Þ
x =
Þ
Lever Rule
Similarly
1-x
Thus,
when one wants to find the average of some quantity such as F, it is usually
sufficient (neglecting surface effects) to take a weighted average.
Consider the free energy F as F(c)
F(c)
= F2 + (F1 – F2) QR/PR
By
similar triangles, Þ F(c ) = SQ in the diagram.
Therefore
one can find free energy of any intermediate composition by drawing straight line between the free energies of the two constituent
phases.
Can
now use this to interpret free energy curves.
Consider
cases of c=0 or negative;
free energy curve had a single minimum.
As pure components (A and B) F(c ) = F
This
can be lowered by going to compositions A1,B1 to give
free energy F1 etc
And as A and B continue to dissolve more and more of each other, free energy
continues to drop.
Minimum
energy for homogeneous single phase, energy Fn
i.e no
phase separation occurs in this case.
Composition
overall determines the state of the mixture.
Minimum
free energy will not be given (in general) by the minimum in the free energy
curve.
e.g. starting with composition c, if this were
to result in composition corresponding to the minimum on the curve cmin, would
necessarily also have phase with composition c' present.
Necessary
condition for homogeneous mixing to occur is for
However
when c is positive, this
inequality does not hold, and the behaviour is very different.
Start
with homogeneous free energy F, for concentration c.
This can split into A1 and B1;
energy drops to F1.
Minimum
in energy occurs at F3 when the representative points on the free
energy curve are joined by the lowest straight line: common tangent construction.
In
this case, have phase separation into compositions cA and cB,
with phases a and b.
For
compositions c < cA have a phase
cA
< c < cB have a + b
cB<c have just b.
Proportions
of a and b given by Lever rule.
For
c<cA A dissolves B
For
cB<c B dissolves A
cA
and cB define solubility limits.
This
common tangent construction can be extended to quite complicated situations,
with several minima, which then give rise to quite complicated free energy
curves and hence phase diagrams.
For
plotting a phase diagram we need to know how solubility limits (as determined
by the common tangent construction) vary with temperature.
Have
seen that if d2F/dc2 everywhere ≥0 have a
homogeneous solution.
Phase
separation occurs when free energy
curve has regions of negative curvature.
This
permits us to evaluate the limits of solubility in terms of c.
For
the symmetric case (i.e FA = FB i.e terms involving EAA
and EBB are assumed equal)
Fmix/
kT = c ln c + (1-c) ln (1-c) + cc (1-c) per site
Critical
value when d2F/dc2 = 0
For
a regular solution c µ 1/T = A/T
Then
limits of solubility vary with temperature according to
i.e.
as T raised the two compositions of phases a and b
converge, until at Tcrit = A/2 there is no further separation and a
homogeneous mixture results.
In
general, critical value of c =
2.
There
is no solution for c for c
< 2 so there is no phase separation.
Also,
for the symmetrical case, the common tangent construction reduces to the
condition
df/dc = 0 (i.e. horizontal tangent)
This
equation defines the binodal or coexistence curve.
For
the regular solution case, with cµ 1/T this allows us
to plot out how the binodal behaves.
Some
comments on this Approach
This
approach of a 'symmetrical' AB mixture is very similar to the Ising model,
where the 2 states correspond to opposite spins (although of course A cannot
transform to B as spins can).
The
approach used has been a mean field theory.
This
will fail:
Þ
near a critical point: will give the wrong critical point and the wrong
critical exponents. In practice most of the phase diagram will be well away
from this point, so this is not too severe.
Þ
for strongly negative c. In this case there are strong attractions
between unlike atoms and the idea of random distributions breaks down. This can lead to order-disorder transitions.
In
general it is difficult to calculate c from 1st
principles.
Picture
described here works best for liquid-liquid phase separation. Melting may complicate matters.
For
solid-solid transitions have additional problems, leading to very complicated
phase diagrams, due to
Strain energy
Other intermediate compounds eg AB2.
However
for the regular solution model we now can construct a phase diagram – which
contains all the essential physics.
c<2 , only a single minimum Þ homogeneous mixture
T decreasing
c>2 , two minima develop Þ phase separation
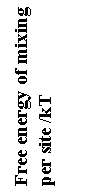
concentration
Knowing the form of the free energy curves as a function of T means that we can map out the limits of solubility etc.
concentration
F
Plotting out the locus of the
binodal enables us to see the shape of the phase diagram.
The
binodal or coexistence curve describes the limits of solid solubility.
Have
seen condition for homogeneous mixing everywhere is d2f/dc2
≥ 0 everywhere.
The locus
d2f/dc2 = 0 is called the spinodal curve.
We
need to distinguish how phase separation occurs inside this curve from between
binodal and spinodal.
Spinodal Curve and Spinodal
Decomposition
In the two phase region, two kinds of decomposition can occur.
1. Nucleation and Growth – which we have already talked about, and is the most familiar.
2. Spinodal Decomposition
In the nucleation and growth regime we know that a nucleus of a critical size has to form before it is energetically favourable for it to grow.
Nucleation and growth corresponds to a metastable region of the phase diagram.
Spinodal decomposition occurs when any composition fluctuation is unstable – the unstable region of the phase diagram.
Section of free energy curve:
Start
at composition c, split into 2 slightly different compositions c1 and
c2 to give net lowering of
energy.
Where
the curvature is negative, any composition
fluctuation leads to a drop in F, and is
therefore unstable.
This
contrasts with region between spinodal and binodal:
Net
increase in F as compositions diverge – hence nucleation barrier, even though
ultimately end up with overall lowering of energy when split into phases a
and b.
Since
any composition fluctuation is stable in the spinodal region, which wavelength
dominates?
Large
fluctuations grow comparatively slowly because atoms
have to diffuse over large distances.
Small fluctuations are suppressed, because they involve a lot of diffuse interfaces.
However
the fact that interfaces aren't sharp means that we have to think carefully
about how to account for the interfacial energy.
Local
free energy density depends on the composition at that point and in the
vicinity, since this will determine the sharpness of the interface.
Local
free energy therefore can be written (1D case)
where A is a constant
Expand
as Taylor series
Discarding
higher order terms, and noting that k11
must be zero by symmetry
Which
can be shown to lead to
Where
K is the gradient energy coefficient
This
theory is due to Cahn and Hilliard for metal alloys. An equivalent theory for magnetic
domains is due to Landau and Ginsburg.
System
will try to equilibrate by having a
uniform chemical potential.
General
transport equation, flux of A, JA
M
is an Onsager coefficient
µA
and µB are the chemical potentials of A and B respectively.
(µA
- µB) is the free energy to remove 1 atom of A and replace it
by B atom
For
early times, c will not differ greatly from overall concentration co. Can then make assumptions M, ¶2fo/¶c2 and K are independent of c, i.e. linearise
– linear Cahn-Hilliard theory.
Now,
continuity equation is
=
And
if the term involving K, the gradient energy term, can be neglected, this
equation is equivalent to a standard diffusion equation with an effective
diffusion coefficient Deff given by
Note
that this diffusion coefficient now contains thermodynamics, and is not simply
a transport coefficient.
M
contains transport information and is always positive.
But
we know that within the spinodal regime ¶2fo/¶c2 is always negative, by definition.
Thus
Deff is negative – 'uphill
diffusion'.
All
fluctuations tend to encourage concentration
fluctuations to increase, so that
neighbouring regions get more and more different.
This
is in contrast to most diffusion.
The contrast between Spinodal Decomposition and
Nucleation and Growth.
Now
return to the full equation involving the gradient energy term.
General
solution to the diffusion equation is
For
equation [1] this becomes
if
the intitial composition is co, where fo" is the
second derivative of fo wrt composition.
This
form of the equation enables us to see which wavelengths grow and which shrink.
The
amplitude of the composition fluctuation of wavevector q changes with an
amplification factor
Recall
fo" is always negative
The
fastest growing wavelength is given by
What does this Morphology look
like?
For nucleation and growth we are used to seeing droplets of one phase nucleating in the other.
For spinodal decomposition typically see (e.g. in an optical microscope) a random, interconnected structure, with a characteristic
lengthscale related to qmax.
From such images, difficult to get a good measure of the average lengthscale.
Easier to do this using scattering methods from which qmax can be directly obtained.
Scattering shows a broad Bragg-like peak, from which qmax can be read off.
The scattering radiation can be light, X-rays… depending on actual lengthscale.
Light scattering works well for polymers, because lengthscales are large, but for atomic species X-rays need to be used as much finer structures.
This simple linearised theory will only work for early times, during which time qmax will be constant.
As time increases non-linear terms come into play, and the analysis gets more complicated.
Domain size R(t); Interface width w(t)
Early R(t)~w(t) Both increase Intermediate R(t)≠w(t) R(t) increases w(t) decreases Late stage w(t) = w(¥)
constant
At early times, linear theory OK; one lengthscale (corresponding to qmax)
Intermediate times – analysis very complicated. Two lengthscales important
Size of domains
Width of interface
Late times – simple theory again with only one lengthscale (interfaces are sharp). Compositions have now reached those of coexisting phases; coarsening continues to occur.
During late stage growth, large droplets grow at the expense of small ones.
Morphology may cease to be interconnected, and starts to resemble that of nucleation and growth regime.
This phenomenon of coarsening also occurs in the nucleation
and growth regime, and is known as Ostwald
ripening.
Why do Small Droplets Shrink and
Large Ones Grow?
Imagine adding one atom from an ideal solution to a precipitate particle, which thereby grows by Dr.
(Same basic argument applies in both metastable and unstable regimes.)
DF = 8πrgDr + DFv4πr2Dr - kT ln c
surface energy bulk free energy entropy change
change change
At equilibrium, DF = 0.
Put W = 4πr2Dr
Then
Now
The local concentration depends on particle size.
Concentration highest just
outside small particles, and the atoms
will diffuse away to try to recover c¥.
Hence small particles shrink,
whilst overall volume fraction remains essentially constant (otherwise c¥ would be changing).
n(r ) = no/unit volume with radius r
n(r ) = no/unit volume with radius r
Can work out how particle size
scales with time by a heuristic argument.
Flux µ chemical potential gradient across interface
Flux also determines how radius grows or shrinks
Þ r3 µ t or
r µ t1/3 Lifshitz-Slyozov
law
This is found to give good agreement with experiment when diffusion, rather than hydrodynamics, is dominating.
Utilising Phase Diagrams
Phase diagrams are inherently for equilibrium.
In practice equilibrium may not always pertain, and one can end up with structures which are far from equilibrium but are 'frozen in' because there is insufficient thermal energy to allow further transformation to occur.
Thus must always bear in mind kinetics as well as thermodynamics.
Case Study I – Zone-refining
Silicon
Silicon chips are only
possible because native silicon, which naturally contains sufficient impurities
to mess up any device, can be purified by a method known as zone-refining.
Starting with co,
when this is melted will be in equilibrium with a solid with concentration kco
(k<1) level of impurities present.
Thus the process of melting and solidification
leads to rejection of impurities into remaining liquid, whose impurity
concentration goes up.
By passing a heater over a
solid bar, can effectively push impurities along.
By doing this several times,
end up with very pure Si (at one end).
Case Study II – Hardening of
aluminium by copper
In this case exactly what
happens, depends on thermal history – e.g. cooling rate, ageing etc.
At the so-called eutectic
point Te, 3 phases are in equilibrium: L, a
and q.
No degrees of freedom.
Corresponds to the composition with the lowest melting point.
At Te, upon
cooling, all remaining liquid solidifies isothermally.
~4% Cu in AL corresponds to
Duralumins, designed with Cu present to harden Al, which is typically a fairly
soft material.
Above 500˚C 4% alloy is
single phase a.
Below 500˚C q
(CuAl2) phase starts to form.
Proportion dependent on T and given by Lever Rule.
However how the microstructure
develops depends on rate of cooling.
Cool slowly – low driving force for nucleating CuAl2
precipitates.
Few nuclei, but these then grow quite large.
Large, well-separated precipitates, typically at grain
boundaries, which do not provide much barrier to dislocation motion
Cool fast – large driving force for nucleation, and therefore
many nuclei form which then do not grow much.
Much finer structure - more
obstacles to dislocation motion
Material which has been fast
cooled is much harder.
However if you cool too fast,
can kinetically frustrate this process, and Cu remains in supersaturated a
solid solution.
Harden now by 'age-hardening'
i.e. hold at low T (typically around 150˚C) for long time (~100 hours).
Fine precipitates will now
form during this heat treatment, by a series of steps.
1. Solid solution – Cu
randomly distributed on lattice.
2. Small 'Guinier-Preston' zones form – small discs of Cu form on 001 Al
lattice planes.
3. These zones coarsen and form q" phase, which
has coherent lattice wrt Al lattice.
This gives rise to 'coherency strain' which strain field impedes dislocation motion. In this case size of lattice mismatch and
distances involved mean that lattice planes can still bend to accommodate this
mismatch.
4. Further coarsening and formation
of q', which is
incoherent (on edges) with Al lattice.
5. Final q phase forms, mainly nucleating at grain boundaries i.e.
heterogeneous nucleation. Because completely incoherent, shape now not
determined by lattice.
Alloy hardest when q"
phase present, since particles have to
cut through precipitates.
Later stages is overaged, and
the large q precipitates provide little obstacle, as dislocations
can bow round the large precipitate particles.